AG Sticht
Professur für Bioinformatik
Prof. Dr. Heinrich Sticht
Kontakt

Forschungsinteressen
Für die Weiterleitung von Informationen in biologischen Systemen spielen Protein-Protein-Wechselwirkungen eine zentrale Rolle. Die Identifizierung und Beschreibung der zugrunde liegenden Prinzipien der molekularen Erkennung mittels computergestützter Methoden trägt wesentlich dazu bei, Regulationsmechanismen zu verstehen und neue, biologisch relevante Proteininteraktionen vorherzusagen. Die Arbeitsgruppe Bioinformatik verwendet bei der Untersuchung derartiger molekularer Wechselwirkungen eine Kombination verschiedener computergestützter Methoden (z.B. Sequenzdatenanalyse, Molekülmodellierung und Moleküldynamik).
Moleküldynamik-Simulationen ermöglichen eine Untersuchung des dynamischen Verhaltens von Proteinstrukturen. Mit ihrer Hilfe können z.B. die Bewegungen viraler Proteine, konformationelle Umlagerungen in menschlichen Proteinen wie dem Alzheimer Aβ-Amyloid und der Einfluss von kovalenten Proteinmodifikationen auf molekulare Erkennungsprozesse untersucht werden. Molekülmodellierungen werden eingesetzt, um die Strukturen von isolierten Proteinen oder biomolekularen Komplexen aufzuklären. Diese bilden die Basis für ein molekulares Verständnis der Effekte von Mutationen auf Proteinstabilität und Bindungseigenschaften. Außerdem werden sequenzbasierende Methoden für eine verbesserte Detektion von funktionalen, linearen Sequenzmotiven entwickelt. Solche Motive spielen eine wichtige Rolle für die Wechselwirkung von zahlreichen Pathogenen mit den Zielmolekülen ihres Wirtsorganismus.
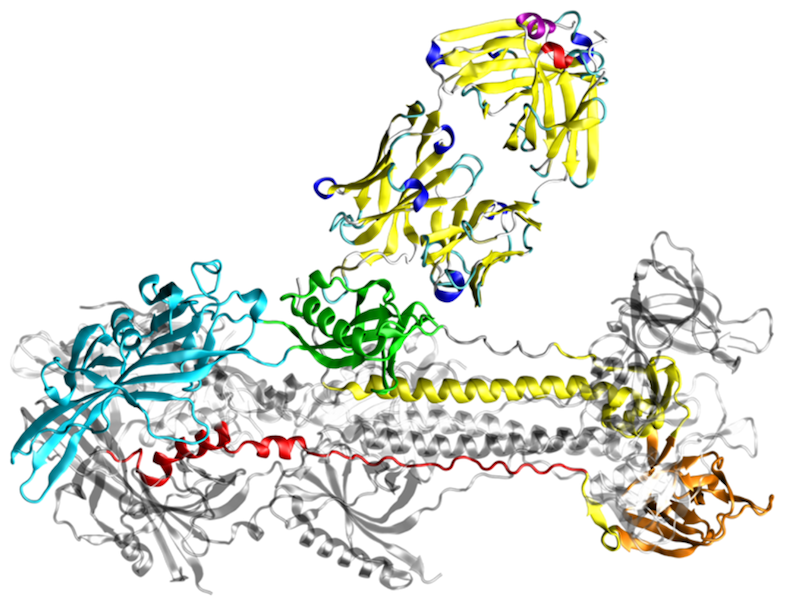
Für Infektionsprozesse spielt die spezifische Interaktion von Pathogenen mit Wirtsproteinen eine wesentliche Rolle. Unsere Forschung konzentriert sich auf die Vorhersage und strukturelle Charakterisierung der Proteininteraktionen zwischen Wirt und Pathogenen mit Hilfe von computergestützten Methoden.
Für die meisten Fragestellungen setzen wir eine Kombination aus Molekülmodellierung, Docking und Moleküldynamik-Simulationen ein. Die letztgenannte Technik liefert Informationen über konformationelle Stabilität und Interaktionsenergien, die sich nicht aus der statischen Struktur ableiten lassen. Unsere Arbeitsgruppe nutzt solche Simulationen zum Beispiel für die Identifizierung und Optimierung von Peptiden, die an virale Fusionsproteine von HIV-1 oder SARS CoV-2 binden und so die virale Infektion blockieren. Einen weiteren Schwerpunkt bildet die Untersuchung der Interaktion von herpesviralen Proteinen mit deren zellulären Bindepartnern.
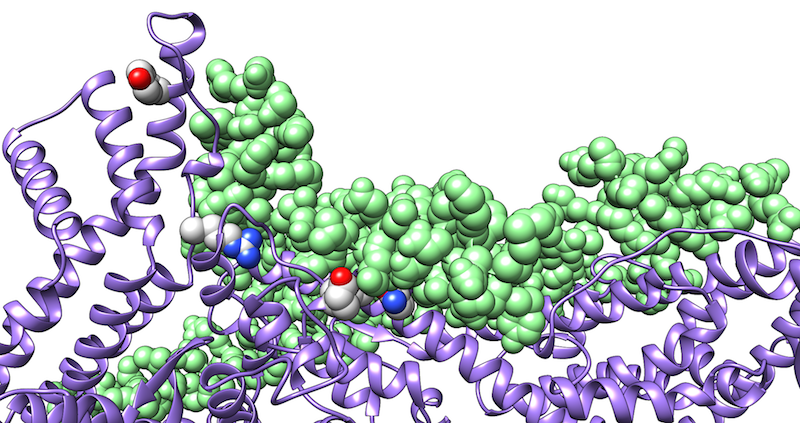
Proteinfehlfaltungserkrankungen sind einzigartig, da sie durch eine drastische Änderung der dreidimensionalen Proteinstruktur hervorgerufen werden. Häufig beinhaltet diese dauerhafte Änderung der Proteinstruktur die Umwandlung einer löslichen, α-helikalen Struktur in eine unlösliche β-Faltblatt-Konformation. Zwar haben Zellen Mechanismen zur Eliminierung dieser unlöslichen Ablagerungen entwickelt; sind jedoch diese Eliminierungsmechanismen überlastet, lagern sich die fehlgefalteten Proteine in Form von unlöslichen, intrazellulären Einschlüssen oder extrazellulären Plaques ab. Solche Ablagerungen fehlgefalteter Proteine sind häufig ein typisches Kennzeichen neurodegenerativer Erkrankungen.
Die Alzheimer-Krankheit als häufigste neurodegenerative Erkrankung ist durch extrazelluläre Proteinablagerungen des Amyloid-Aβ-Fragments (Aβ) und durch intrazelluläre Tau-Filamente, sogenannte neurofibrilläre Bündel, gekennzeichnet. Die räumliche Struktur der Aβ-Ablagerungen zeigt zwar die typische Topologie von Fibrillen, enthält aber nur wenige Informationen über die Rolle der einzelnen Aminosäurereste für die Fibrillenbildung. Diese Informationen sind jedoch wichtig für die Entwicklung neuartiger Medikamente, die Aβ-Aggregation verhindern oder die gebildeten Aggregate auflösen, indem sie an Schlüssel-Aminosäuren binden, diese abschirmen und dadurch die fibrilläre Struktur beeinflussen. In diesem Zusammenhang führen wir Moleküldynamik-Simulationen von Aβ-Oligomeren und thermodynamische Analysen der Interaktionsflächen innerhalb der Aβ-Aggregate durch. Darüber hinaus untersuchen wir die Wirkung verschiedener Lösungsmittelumgebungen auf die konformationelle Stabilität dieser Aβ-Oligomere.
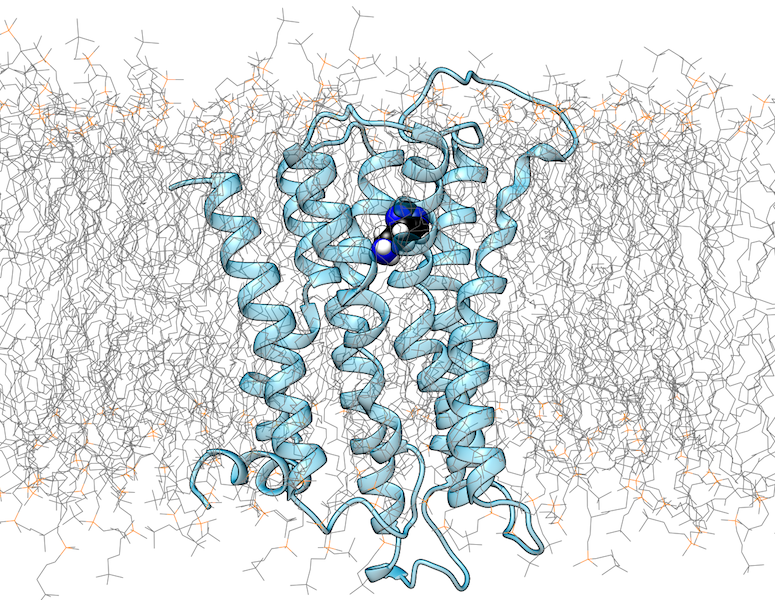
G-Protein gekoppelte Rezeptoren (GPCRs) sind membranständige Proteine, die extrazelluläre Liganden erkennen und dadurch intrazelluläre Signalprozesse auslösen können. Inzwischen existieren mehr als 100 GPCR-Kristallstrukturen, die einen wertvollen Einblick in die strukturellen Eigenschaften dieser Proteinfamilie geben. Allerdings ist das aktuelle Verständnis der Struktur und Funktion von GPCRs noch alles andere als vollständig.
Ein wesentlicher Grund dafür ist, dass GPCRs in aktiven und inaktiven Konformationen vorkommen, von denen meistens nur ein Zustand kristallisiert werden konnte. Darüber hinaus wird die Struktur durch die Interaktion mit intrazellulären Bindungspartnern (IBPs) wie G-Proteinen oder β-Arrestin beeinflusst. Auch die extrazelluläre Liganden-Bindetasche weist eine gewisse strukturelle Plastizität auf, was insbesondere für die Wirkstoffentwicklung relevant ist. Da eine Kristallisation von GPCRs in verschiedenen Aktivierungszuständen oder im Komplex mit verschiedenen Liganden experimentell sehr aufwendig ist, kommt computergestützten Methoden für die Untersuchung dieser Aspekte eine herausragende Bedeutung zu.
Wir nutzen Methoden der Molekülmodellierung und Moleküldynamik, um die Struktur von GPCRs im Komplex mit verschiedenen niedermolekularen Liganden oder intrazellulären Interaktionspartnern zu studieren. Untersuchte Fragestellungen beinhalten die Vorhersage der Bindungsmodi niedermolekularer Liganden, konformationelle Änderungen in GPCRs als Folge der Ligandenbindung, sowie den Einfluss von Mutationen auf GPCR-Funktion und -Interaktion. Zusätzlich zu konventionellen MD-Simulationsmethoden werden dabei auch rechnerisch aufwendige Metadynamik-Simulationen eingesetzt.
Neben GPCRs untersuchen wir auch andere Klassen von membranständigen Rezeptoren mit ähnlichen methodischen Ansätzen. Dazu gehört der Glycin-Rezeptor, an dem wir die Bindestelle von Sachcariden als allosterische Modulatoren charakterisieren. Im Fall des Macrophagen-Oberflächenrezeptors Mincle untersuchen wir die Bindung synthetischer Glykolipide, was langfristig die Entwicklung besserer Adjuvantien für Impfstoffe unterstützen soll.
Änderungen des pH-Werts regulieren viele biologische Prozesse in Bakterien, Viren, Wirbeltieren und Pflanzen. So können zum Beispiel einige Bakterien die sauren Bedingungen im Magen ihres Wirts mit Hilfe von säure-aktivierten Chaperonen überleben, die ihre Substratproteine vor Aggregation schützen. In manchen Viren existieren pH-abhängige Fusionsproteine, die den Eintritt in die Zelle vermitteln. Proteine in Wirbeltieren erfahren z.B. pH-Unterschiede auf dem Weg durch das Endoplasmatische Reticulum und den Golgi-Apparat. Um die Situation experimenteller Titrationsexperimente nachzubilden untersuchen wir pH-abhängige Proteine mittels Moleküldynamik(MD)-Simulationen, in denen der pH-Wert über die Zeit variiert wird. Diese Methode erlaubt die Berechnung von Titrationskurven und den pKa-Werten ionisierbarer Gruppen. Mit dieser Strategie untersuchen wir auf atomarer Ebene den Effekt von pH-Änderungen auf die lokale Proteinstruktur, die Interaktionseigenschaften und die konformationelle Stabilität.
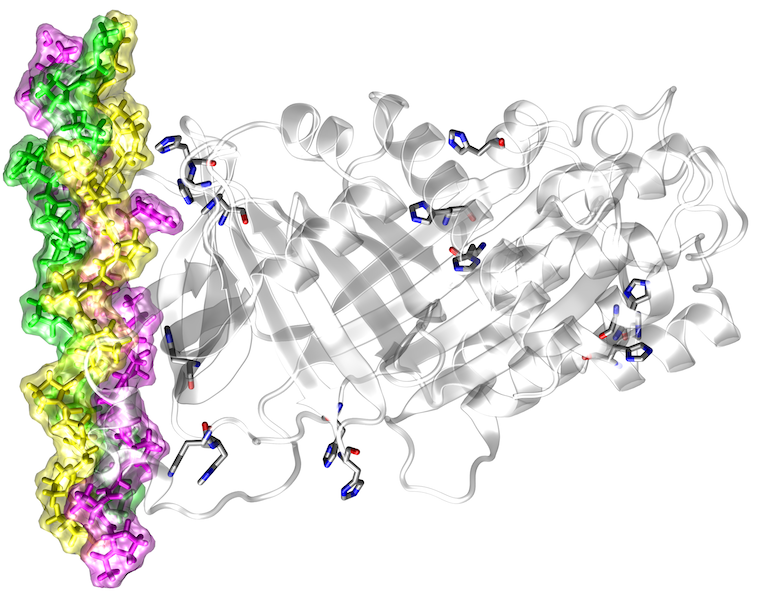
Hochdurchsatz-DNA-Sequenzierungsstudien haben gezeigt, dass es eine hohe genetische Variabilität zwischen Individuen gibt. Viele dieser Sequenzvarianten führen zu Aminosäureaustauschen, von denen einige mit Krankheiten in Zusammenhang stehen. Aufgrund ihrer großen Anzahl (> 10.000 pro Genom) ist es unmöglich, alle Sequenzvarianten experimentell zu charakterisieren, so dass rechnergestützte Vorhersagewerkzeuge für die Identifizierung pathogener Varianten von größter Bedeutung sind. Die meisten bisherigen Methoden verwenden evolutionäre Konservierung und andere sequenzbasierte Merkmale, um schädliche Varianten zu identifizieren, aber sie können die Auswirkungen dieser Varianten auf die Proteinfunktion nicht vorhersagen. Trotz ihres unmittelbaren Bezugs zur Proteinfunktion werden Strukturinformationen in den Vorhersagen derzeit nur sehr begrenzt berücksichtigt. Darüber hinaus konzentrieren sich die wenigen bestehenden strukturbasierten Vorhersagemethoden hauptsächlich auf einen bestimmten Aspekt der Proteinstruktur (z.B. Proteinstabilität oder Proteininteraktionen) und erlauben daher keine umfassende strukturelle und funktionelle Annotation. Ziel des aktuellen Projekts ist die Entwicklung eines robusten Frameworks für eine umfassende strukturbasierte Analyse und Interpretation von Hochdurchsatz-Sequenzierungsdaten.